2024-2025 幸运飞行艇-官方5分钟飞艇168开奖结果-官网开奖记录查询 Annual Report
The power of networks
2023 highlighted how important networks are in biology and research, driving progress in biomedicine.
This report explores the interconnectedness that fosters innovation, from cellular systems to the patient, covering ambitious scientific collaborations with top research centres, companies, and hospitals.
2023 in numbers

79.7%
PUBLICATIONS IN D1 JOURNALS

€34.1M
BUDGET
* Provisional data

28
RESEARCH GROUPS
Research

177
SCIENTIFIC PUBLICATIONS
In 2023, IRB Barcelona has once again stood at the forefront of biomedical research, making pivotal advancements in understanding and combating cancer and metastasis, as well as tackling the complex challenges of ageing and metabolism-related disorders. Leveraging the power of extensive research networks, we've deepened our exploration into the intricate web of interactions underpinning health and disease.
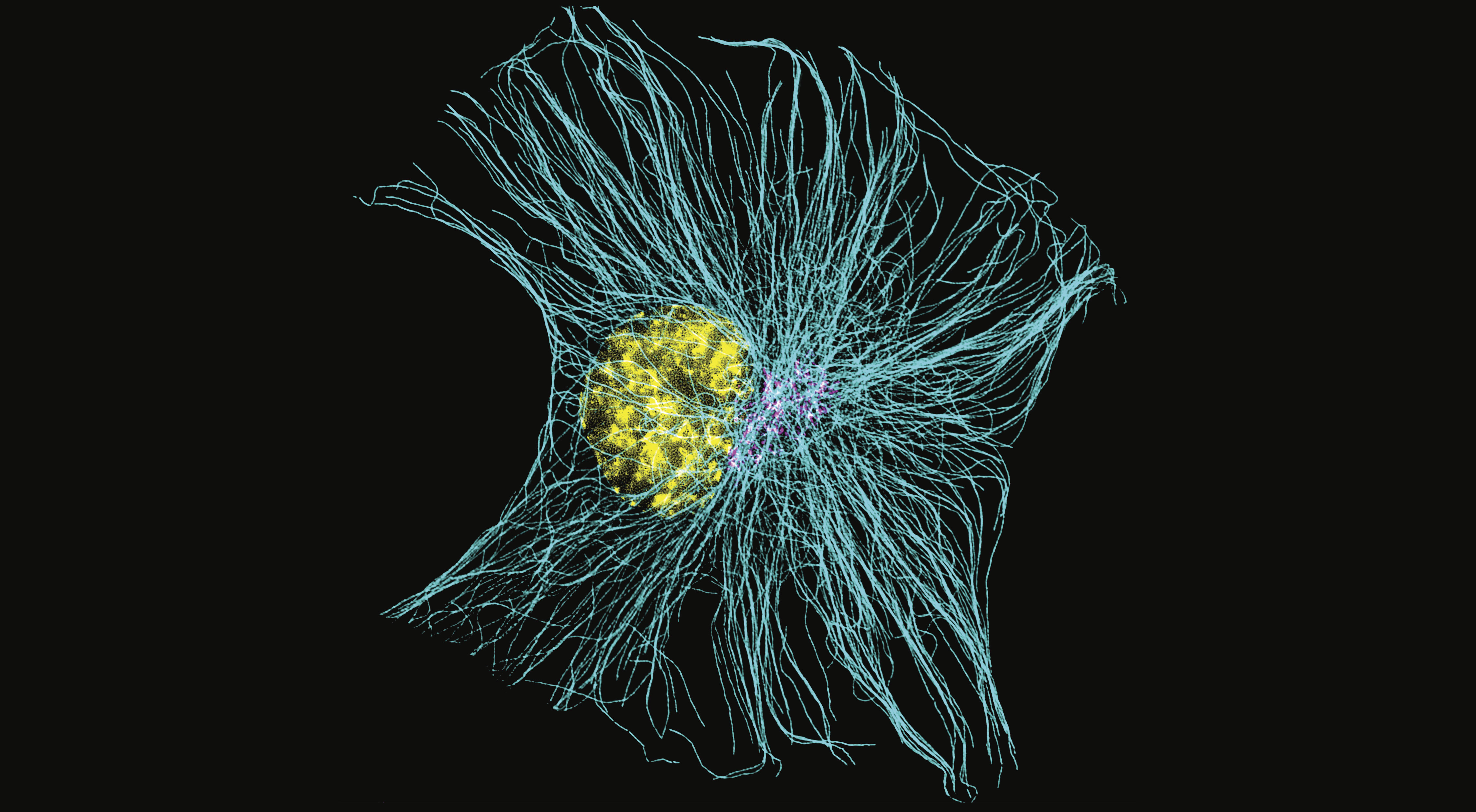
Published in Nature Cell Biology
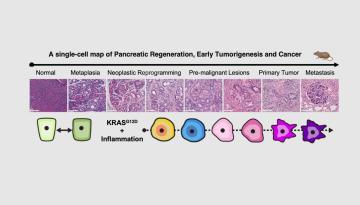

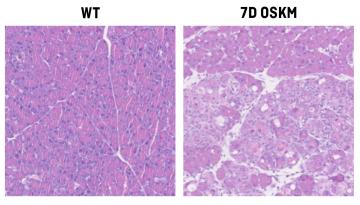
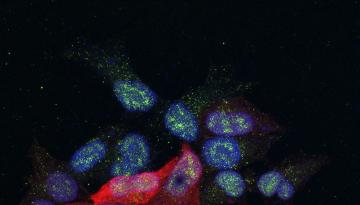
Published in Nature Metabolism
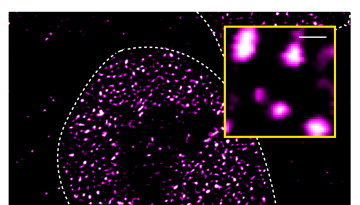
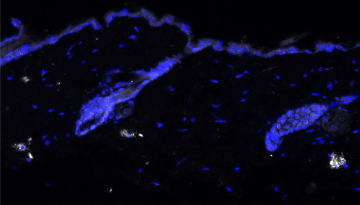
Published in Nature Structural & Molecular Biology
Scientific projects

€19.8M
EXTERNAL FUNDING
In 2023, IRB Barcelona has been distinguished by the successful acquisition of funding for a range of groundbreaking projects through both competitive and non-competitive grants offered by various public and private entities.





Innovation

6
ACTIVE SPIN-OFFS
This year has seen unprecedented breakthrough with one of our spin-offs launching its first biomedical diagnosis tool into the market. Another spin off has secured substantial seed funding. These developments highlight our dedication to turning research into clinical applications, showcasing our impact on both the biotech industry and patient care.
The test is available to oncologists and pathologists and will benefit an estimated 24,000 patients each year in…
Nuage Therapeutics, a spin-off of IRB Barcelona and ICREA, has raised €12M in Seed Financing to advance its lead…
The IRB Barcelona spin-off Gate2Brain has won the Senén Vilaró Prize for pioneering technology facilitating drug…
Dr. Toni Gabaldón and Dr. Salvador Aznar Benitah have been awarded ERC Proof of…
People & Talent

524
PROFESSIONALS




Communications & Public Engagement

3,524
Impacts in media
In 2023, our research has significantly increased our media presence, attracting attention from specialist journals, mainstream media, and digital platforms, underscoring our scientific impact and global reach.

4,641
PARTICIPANTS IN IN-PERSON OUTREACH ACTIVITIES
We have reached an extensive audience through our outreach initiatives, in line with our long-standing commitment to nurturing scientific curiosity among the next generation and engaging the wider community in the wonders of science.

Fundraising

€3M
Raised since the launch of the Metastasis Challenge
The Metastasis Challenge has evolved beyond a fundraising campaign into a dynamic movement that rallies individuals and organisations together to combat metastatic cancer. This collective drive has successfully mobilised resources, amassing over €3M, underscoring the strength of unity in facing the challenges posed by metastatic cancer, and demonstrating how interconnected efforts can lead to substantial progress in the quest for a cure.

Thank you!
We extend our gratitude to all the individuals and institutions that have contributed to making 2023 a remarkable year.
We are also profoundly grateful to every person and organization that has supported the Metastasis Challenge.
Every donation, every event, every legacy helps us get one step closer to our challenge of stopping metastasis.